O QUE É A SINDROME DE HUGHES?
A Síndrome de Hughes, também chamada Síndrome Antifosfolípido, faz parte do extenso grupo de doenças auto-imunes, em que os mecanismos de defesa, que protegem habitualmente o indivíduo, estão desregulados, pelo que começam a produzir substâncias – auto-anticorpos – que agridem várias estruturas do próprio organismo.
Na Síndrome de Hughes a produção de anticorpos está associada a um excesso de coagulação do sangue e ocorrência de tromboses.
Esta síndrome é conhecida por Síndrome de Hughes, porque foi o Professor Graham Hughes e os seus colaboradores que, em 1983, em Londres, identificaram como uma nova doença a associação de tromboses arteriais e venosas recorrentes em vários órgãos e sistemas, abortos de repetição e a presença de anticorpos antifosfolípido em análises de sangue.
A Síndrome de Hughes pode existir isolada (forma primária, a mais frequente), ou associada a outras doenças auto-imunes como, por exemplo, o Lúpus (forma secundária).
QUE PESSOAS SÃO MAIS AFECTADAS?
A Síndrome de Hughes atinge igualmente o sexo masculino e feminino, muitas vezes em idades jovens. Na mulher, o primeiro episódio pode ocorrer com o início do uso da pílula.
QUAIS AS MANIFESTAÇÕES MAIS FREQUENTES?
TROMBOSES
As manifestações mais frequentes resultam da tendência aumentada para tromboses, que podem ocorrer em qualquer ponto do organismo, em grandes ou pequenos vasos da circulação arterial ou venosa.
A trombose venosa dos membros inferiores é a mais comum, acompanhando-se de dor intensa e “inchaço” (mais raramente nos membros superiores). Algumas vezes acompanha-se de embolia pulmonar. Pode também ocorrer em veias de órgãos internos – fígado, rim, cérebro, olho, etc.
A trombose arterial é mais grave, mas menos frequente. Atinge mais vezes os grandes vasos cerebrais, provocando isquémia (morte de céluas por falta de irrigação sanguínea), mas pode atingir os vasos cerebrais mais pequenos, podendo resultar em demência. Pode ocorrer também confusão mental de curta duração, enxaqueca, epilepsia, perda de memória ou alterações temporárias da fala.
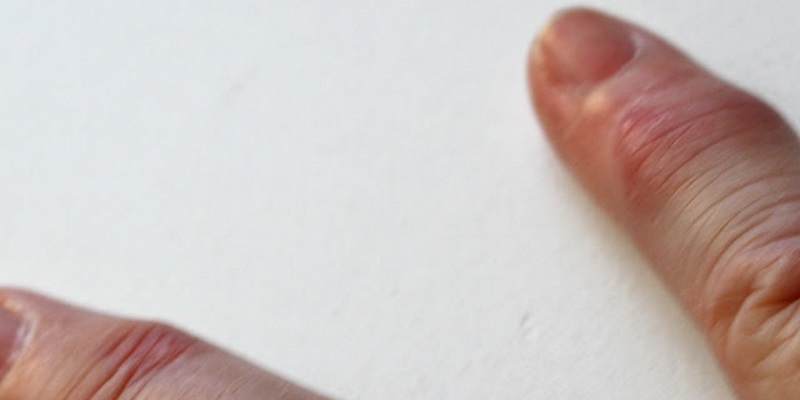
A isquémia em outros órgãos pode provocar complicações diversas: enfarte do miocárdio (coração); insuficiência renal aguda ou hipertensão grave (rim); dor abdominal súbita intensa (intestino); marcha dolorosa ou úlcera de perna crónica (membros inferiores); manchas azuladas, em forma de rede, nas extremidades dos membros superiores e inferiores, que sofrem agravamento pelo frio (livedo reticular); dor e perda súbita da visão.
INSUCESSO OBSTÉTRICO
Durante a gravidez ocorrem alterações fisiológicas no organismo, sendo uma delas o aumento da viscosidade sanguínea (sangue espesso). A grávida com Síndrome de Hughes está mais exposta ao risco de trombose, que resultam em enfartes da placenta.
O insucesso obstétrico pode apresentar-se das mais variadas formas: dificuldade em engravidar, abortos de repetição, perdas fetais, atraso de crescimento intra-uterino ou infertilidade.
Em muitas mulheres, a única manifestação da Síndrome de Hughes, é o insucesso obstétrico, apenas desenvolvendo a doença durante a gravidez.
É importante saber que, sob tratamento adequado, é possível obter uma percentagem importante de gravidezes bem sucedidas.
DIMINUIÇÃO DAS CÉLULAS SANGUÍNEAS
A trombocitopenia ou diminuição do número de plaquetas (células reguladoras da coagulação) é a manifestação mais frequente da doença, mas raramente tem gravidade e quase nunca se manifesta sob a forma de hemorragias.
Pode também acontecer uma anemia hemolítica, em que há uma destruição de glóbulos vermelhos, o que pode provocar uma anemia importante.
Estas manifestações raramente são graves, sendo o tratamento feito com cortisona, aspirina ou heparina.
COMO É FEITO O DIAGNÓSTICO?
O diagnóstico é baseado nas manifestações clínicas e em critérios laboratoriais.
Nas primeiras incluem-se tromboses venosas ou arteriais recorrentes e/ou insucesso obstétrico, com um ou mais abortos, nascimentos prematuros e morte fetal.
Nos critérios laboratoriais incluem-se a presença de anticorpos antifosfolípido em análises de sangue, anticorpos anticardiolipina (A.C.A.), anticoagulante lúpico (L. A.) e antiß2glicoproteína1 (antiß2GP1).
Por vezes, os anticorpos estão presentes em outros tipos de doenças como tumores, infecções, ou em doentes a fazer medicamentos de utilização comum, sem, no entanto, serem causadores de doença trombótica.
QUAL É O TRATAMENTO?
É importante saber que existe tratamento e que este é eficaz em muitas situações.
Não se deve esquecer, no entanto, que há sempre um risco aumentado de tromboses em relação ao indivíduo saudável.
Os medicamentos mais utilizados são os anti-agregantes, como a aspirina em doses baixas (100 mg/dia) e os anticoagulantes, como a varfarina e a heparina, com o objectivo de tornar o sangue mais fluido, impedindo a trombose vascular.
A gravidez é sempre considerada de risco e deve ser programada e fortemente vigiada pelo médico assistente habitual e pelo obstetra.
Nesta situação é preconizada a utilização da aspirina, que deve ser iniciada quando da programação, mas pode ser necessário associar a heparina injectável (sem praticamente risco para a mãe ou para a criança), sobretudo em situações em que a aspirina não foi bem sucedida anteriormente. A percentagem de sucesso passa de 20% para 70% apenas com este tratamento!
A varfarina deve ser evitada na gravidez, porque afecta o desenvolvimento do feto.
Nas manifestações cerebrais ligeiras como a enxaqueca, perturbações temporárias da fala ou epilepsia, a aspirina ou a varfarina também são eficazes.
Nas tromboses venosas e arteriais recorrentes e graves a varfarina é o anticoagulante utilizado e vai ser necessário durante toda a vida. O seu médico vai periodicamente pedir uma análise – I.N.R. (deve estar perto do valor 3) que serve para ajustar a dose de medicação.
O excesso de anticoagulação pode provocar hemorragias, pelo que é importante ser vigiado.
A QUEM DEVE RECORRER?
A Síndrome de Hughes exige a colaboração de uma equipe multidisciplinar. Deve haver uma estreita ligação entre o Médico de Medicina Geral e Familiar e os especialistas com experiência neste tipo de doença, sendo frequente haver necessidade da intervenção de outras especialidades, assim como a de outros técnicos como sejam enfermeiros, psicólogos, fisioterapeutas, assistentes sociais e outros.
Os Especialistas com mais experiência neste tipo de doenças são os de Medicina Interna e de Reumatologia.
Os especialistas de Medicina Interna são os médicos do adulto que abordam e tratam os doentes como um todo, recorrendo aos especialistas de determinados órgãos, para a execução de técnicas ou para apoio no tratamento de doenças mais raras desses órgãos ou sistemas.
Essa capacidade torna-os particularmente vocacionados para este tipo de patologias que têm um carácter sistémico, ou seja, podem atingir vários órgãos sucessivamente ou ao mesmo tempo.
Em grande parte dos hospitais existem consultas diferenciadas, chamadas Consultas de Doenças Auto-imunes ou de Imunologia Clínica, dirigidas por internistas, com grande experiência neste tipo de doença.
A lista destas consultas nos hospitais do Serviço Nacional de Saúde é apresentada no verso, ou consulte em www.nedai.org.
Ver documento